You looked up the pKa. You set your mobile-phase pH a comfortable two units away. The method still drifted.
It happens more than people expect, and the reason is quiet. The pKa you looked up is almost certainly not the pKa that was running your separation.
The pKa you looked up is the wrong one
Published pKa values are nearly always the thermodynamic pKa: measured in pure water, at 25 degrees, at low ionic strength. Your reversed-phase mobile phase is none of those things.
The organic is the big difference. Add methanol or acetonitrile and the pKa moves, because forming a charged species is harder in a less polar solvent. The shift is not small. In a hydro-organic mobile phase the apparent pKa can sit a full unit or more from the aqueous value, and the direction depends on the analyte and the organic. A compound with a database pKa of 4.0 in water can behave as if its pKa is 5.5 in fifty percent acetonitrile. Set your pH to 6 on the strength of that 4.0 and you think you are two units clear. You are barely half a unit from the pKa that is actually moving your retention, back on the steep part of the curve.
Temperature moves it too, which is how a validated method can drift when the lab cools. Ionic strength moves it. And for a new compound, an impurity, or a degradant, there is often no literature value at all, only a software prediction with real uncertainty. For a molecule with several ionizable groups, the value you find may even be assigned to the wrong group.
The one that matters is the apparent pKa, in your mobile phase
So the number you actually need is the apparent pKa under your method’s conditions: the same organic, the same buffer, the same temperature. That is the pKa whose transition zone your operating pH has to clear. Anything else is an estimate of where the danger is, measured in the wrong solvent.
The good news is you do not need a titration rig or a separate instrument to get it. You can measure it on the system you are already developing the method on.
Measure it from retention against pH
Run the analyte at several mobile-phase pH values across the range where you expect the pKa, holding the organic, the buffer concentration, and the temperature constant. Five or six points spanning a few units, with finer steps where the retention starts to move. Record the retention factor, k, at each.
Plot k against pH and you get a sigmoid. At low pH one ionization state dominates and retention sits on a plateau. At high pH the other state dominates and retention sits on a second plateau. In between, where the molecule is switching between forms, retention swings. The midpoint of that swing, the inflection of the curve, is the apparent pKa, measured in the exact mobile phase that matters.
Read it off the first derivative
There is a catch. The inflection of a sigmoid is the hardest point on it to locate by eye. The curve is gentle right there, so you end up interpolating a midpoint and guessing.
The fix is the one a reader raised when I wrote about this on LinkedIn, and it is a good one. Take the first derivative of the curve, the change in retention per unit of pH between adjacent points. The inflection, where the curve is changing fastest, becomes a maximum. A peak. And the apex of a peak is something a chromatographer can pin down to a tenth of a pH unit without arguing about it.
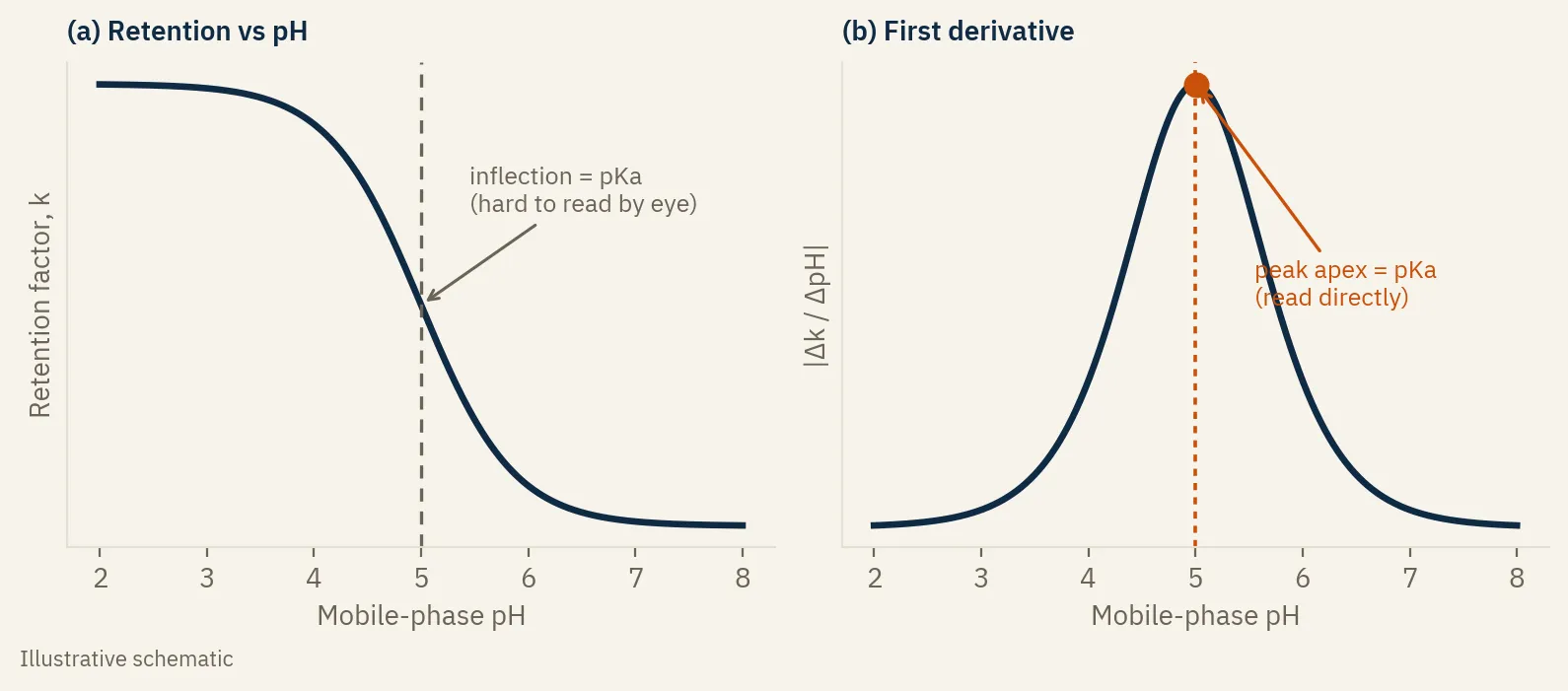
It is the same logic you use to find the endpoint of a potentiometric titration from the first derivative of the pH curve, and the same idea works on a spectrophotometric absorbance-versus-pH curve. It has a bonus: if a molecule has two pKa values close together, two faint bends in one sigmoid are nearly impossible to separate by eye, but they show up as two distinct peaks in the derivative. The same trick locates the isoelectric point of a zwitterion, which sits between its two relevant pKa values.
The same experiment has a second life. Run the analyte across pH alongside a set of reference compounds of known lipophilicity, and the calibrated retention gives you a chromatographic logD at each pH, the lipophilicity profile discovery labs use to judge whether a new molecule is developable. Same data acquisition, a different question. I’ve written about measuring logD that way.
What it buys you
Once you have the apparent pKa in your own mobile phase, the rule you already know finally means something. Keep your operating pH at least two units from it, on the flat part of the curve, and a small drift in pH or a few degrees of temperature barely moves your retention. Sit inside those two units and you have built a method that holds until the column ages, the buffer lot changes, or the lab cools. Then it drifts, and you spend a week finding out why.
flowchart TD
A[Ionizable analyte,<br/>retention matters] --> B[Run k vs mobile-phase pH<br/>on your own system]
B --> C[Plot the sigmoid]
C --> D[Take the first derivative]
D --> E[Read the apparent pKa<br/>off the peak apex]
E --> F[Set the mobile-phase pH<br/>2 or more units from every pKa]
F --> G[Robust by design]I have written before about keeping the mobile-phase pH clear of every pKa. This is the part underneath it: clear of every pKa as your method sees it, not as a database measured it in water.
The point
Measuring a pKa sounds like physical-chemistry homework, the kind of thing you leave to a textbook value. As an estimate of where to look, the textbook value is fine. As the number you set your method on, it is the wrong one.
So when retention matters and the analyte is ionizable, spend the afternoon. Run it across pH on your own system, differentiate the curve, and read the apparent pKa off the peak. Then place your mobile phase a real two units clear of it. That is a method that holds because you built it on the right number.
If you want the short version of the questions to settle before a method gets this far, the validation-readiness checklist is below. Questions about any of this, you can reach me on LinkedIn.