A new molecule lands on your bench with a structure and not much else. Before anyone spends real money on it, someone wants to know whether it will cross a membrane, dissolve, and survive a formulation. A lot of that comes down to one number: how lipophilic it is, at the pH that matters.
That number is logD. The textbook way to measure it, the shake-flask, is the slow way. At a CRO I spent a good stretch measuring logD a different way, on an HPLC, and it changed how fast we could turn a molecule around.
What logD tells a discovery team
logP is a molecule’s lipophilicity in its neutral form: how it splits between octanol and water. logD is the same idea at a specific pH, counting every form of the molecule, neutral and ionized. For anything ionizable that distinction matters, because the charged form barely partitions into octanol while the neutral form does.
Discovery cares because logD at physiological pH, logD 7.4, predicts a lot: membrane permeability, solubility, plasma protein binding, and whether a compound is even worth progressing. Too low and it won’t cross membranes. Too high and it won’t dissolve and sticks to everything. A team screening hundreds of compounds needs that number early, cheap, and fast.
Why the shake-flask is the slow way
The reference method is the shake-flask: equilibrate the compound between octanol and aqueous buffer at a set pH, then measure the concentration in each phase. It works, and it is still the arbiter. For screening it is painful. It needs a lot of compound, which you don’t have early. It is slow to equilibrate, it emulsifies, and it only spans a narrow logD range before the concentration in one phase is too low to measure. Run a few hundred compounds that way and you are the bottleneck.
Measuring logD on an HPLC
Reversed-phase retention is itself a lipophilicity measurement. A compound retains on a C18 column for the same reason it partitions into octanol: it prefers the non-polar phase. So if you run a set of reference compounds of known logD and plot their retention against it, you get a calibration line. Any new compound’s retention then reads back to a logD off that line.
That is the method I ran. Inject the compound and the reference set under fixed conditions, measure retention, convert. It uses micrograms, not milligrams. It runs in minutes. It covers a wide logD range without the compound falling out of one phase. And because it is an HPLC method, it is reliable and easy to automate for a screen.
flowchart TD
A[New molecule + reference standards<br/>of known logD] --> B[Run all on RP-HPLC,<br/>fixed conditions]
B --> C[Measure retention]
C --> D[Calibrate: retention → logD<br/>from the reference line]
D --> E[Repeat across pH]
E --> F[logD-vs-pH profile,<br/>plus logP and the pKa]There are a couple of established flavors. One extrapolates the retention factor to fully aqueous conditions (log k_w). Another uses a fast gradient and a calibrated scale (the chromatographic hydrophobicity index, CHI). Both turn retention into a lipophilicity number you can defend.
logD across pH, and the link to pKa
Run that measurement at one pH and you get a single logD. Run it across several pH values and you get a logD-versus-pH profile, which is where it connects to everything else.
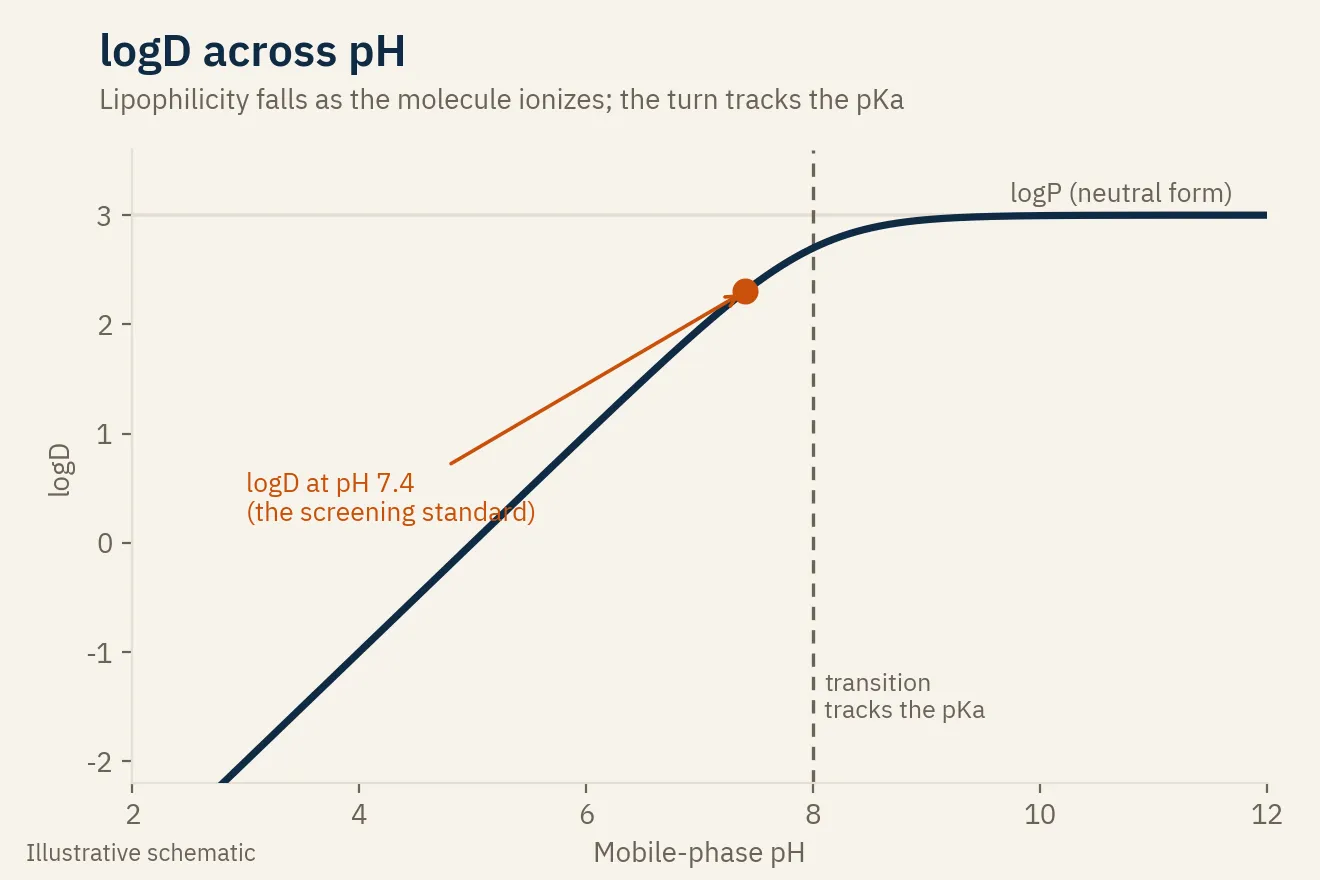
The profile is a curve: a plateau at the logP of the neutral form, dropping away as the molecule ionizes, and the pH where it turns is set by the pKa. So the same kind of retention-versus-pH data that tells you a compound’s apparent pKa also gives you its lipophilicity profile. One screen yields two of the three numbers, pKa, logP, and logD, that decide whether a molecule is developable.
What the method is good for, and where it breaks
A chromatographic logD is a calibrated correlation. It estimates the partition from retention rather than measuring it directly, so it is only as good as the reference set, and it can miss for chemotypes that retain for reasons beyond simple lipophilicity: strong silanol interactions, unusual shape, a logD that sits past the calibration range. The honest way to run it is as a high-throughput screen, with the shake-flask held in reserve to confirm the compounds that matter or the ones that look strange.
Used that way, it is one of the highest-leverage measurements a discovery lab has: a fast, cheap read on developability, early enough to act on.
Why it matters
The molecules that fail late are expensive, and a lot of those failures were visible early, in the physchem. A logD too high to dissolve. Too low to permeate. A pKa that would fight every formulation. Measuring logD fast and early is how a discovery team stops the wrong molecules before they cost real money, and spends its effort on the ones that can actually become a drug.
If your group is standing up or troubleshooting a chromatographic physchem screen, or weighing whether to build it in-house or send it out, that is the kind of thing I help labs work through. Questions, you can reach me on LinkedIn.