I’ve been handed the same kind of problem more than once. A hydrophilic peptide active sitting at low ppm in a finished cosmetic, a matrix full of silicones and surfactants, and a method that doesn’t exist yet. Someone needs a number, and they needed it last week.
The instinct in most labs is to open the solvent cabinet and start running columns. I think that’s usually the slowest way to get there.
Here’s the claim I want to make, and it’s one most labs won’t accept: the fastest path to a validated method is usually to stop running experiments for a while.
Take Acetyl Hexapeptide-8, better known as Argireline, the anti-aging active in a lot of serums. It’s a good worked example, because almost everything that makes peptide method development hard shows up in one molecule. Here’s how I think about it before touching the instrument.
The meeting trap
When a method is late, the problem has a way of migrating into a conference room. Formulation, regulatory, and analytical sit down to agree on a workaround. That’s what happens when something that lives in an instrument gets pushed into a meeting.
Meetings don’t develop methods. They buy time while the samples sit.
I’ll admit the desk-first approach isn’t an instinct I was born with. Early in my career I did what most junior analysts do: I opened the solvent cabinet and started running. Trial and error. Iterative. Expensive. The first two years at the bench taught me that blind runs are mostly a way of documenting your confusion in chromatogram form.
A polar peptide in a cosmetic matrix is one of the clearest tests of the alternative.
Start with the structure, not the column
Acetyl Hexapeptide-8 is a synthetic hexapeptide: Ac-Glu-Glu-Met-Gln-Arg-Arg-NH2. Molecular weight 889.1 Da. Highly hydrophilic, log P of -6.3. The N-terminus is acetylated, the C-terminus amidated. Those modifications eliminate the standard zwitterionic handles most peptide methods rely on for retention.
What you’re left with are four ionizable side chains: two glutamic acid residues (pKa ~4.3) and two arginine residues (pKa ~12.5). The arginines stay positively charged across the entire usable pH range of a silica column. The glutamic acids are pH-dependent.
That told me two things immediately.
A standard C18 reversed-phase method wasn’t going to work. A peptide this polar elutes near the solvent front on C18, which is exactly where the cosmetic matrix dumps all its polar garbage. Testing the peptide in that window means testing it in maximum noise.
But I had a pH handle. By adjusting mobile phase pH to around 5, I could partially deprotonate the glutamic acid residues and shift retention without touching the elution of non-ionizable matrix components like silicones.
That’s time-shifting. Before I touched the instrument, I knew roughly where I wanted the peptide to elute and why.
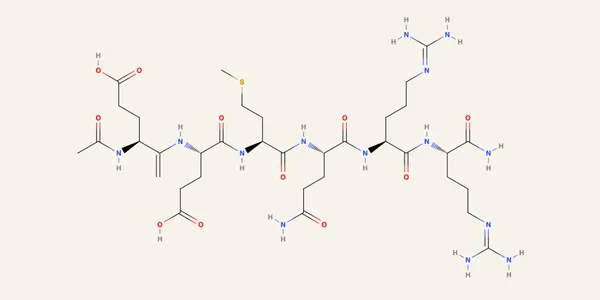
The matrix is the actual problem
The active peptide is present at low ppm in a finished cosmetic. The matrix is everything else: emulsifiers, thickeners, polymers, silicones, hyaluronic acid. In ESI-MS, that matrix kills signal.
Ion suppression in electrospray is a physical process. The ESI source forms a Taylor cone under high electric field, producing charged droplets. As those droplets shrink through solvent evaporation, matrix components compete for surface charge and interfere with ion formation.
The suppression mechanisms in this formulation were distinct for each matrix class, and each required a different response.
Surfactants like PEG-100 Stearate are highly surface-active. They migrate to the outside of the ESI droplet and monopolize available protons. The peptide never gets charged. You see nothing. Carbomer, the thickener, increases droplet viscosity, slowing solvent evaporation and droplet fission, so the analyte gets trapped in large non-volatile clusters that never reach the detector. Hyaluronic acid is a large molecule that physically clogs column particles if you inject crude matrix. I removed it by precipitation with salt and organic solvent before injection. Dimethicone and cyclic silicones can physically coat the peptide and prevent the transition from liquid to gas phase, and they contaminate the ion source over time.
A method that doesn’t account for each of these specifically is a guess dressed up as a protocol.
Why HILIC, and why the Shodex column
I chose HILIC over reversed-phase for one reason that follows directly from the matrix problem: in HILIC, non-polar cosmetic excipients elute fast and the polar peptide elutes late. That’s the opposite of C18 behavior. The peptide ends up in a cleaner window, away from the highest-density matrix interference.
HILIC works by maintaining a water-enriched layer on the polar stationary phase surface. The analyte partitions between the high-organic bulk mobile phase and this aqueous layer. For Acetyl Hexapeptide-8, that means strong retention, and it means I could use a high-acetonitrile mobile phase. High-organic mobile phases improve ESI sensitivity compared to the high-water conditions you’d need for peptide elution on C18. Two problems solved by the same column choice.
I used a Shodex HILIC column with an ammonia-buffered mobile phase at basic pH. Shodex has application notes on similar systems. The basic pH ensures clean peak shape for the arginine-rich peptide. TFA, the obvious choice for a peptide HPLC-UV method, contaminates LC-MS systems and causes ion suppression. It was never an option here.
Choosing the instrument parameters
On a Waters Xevo TQ-S, the instrument choice mattered.
The StepWave ion guide in the TQ-S deflects neutral species away from the ion path before they reach the high-vacuum region. In a dirty cosmetic matrix, that’s significant. Neutral contaminants from silicones, polymers, and matrix residue get excluded early, which improves signal-to-noise and keeps the source clean longer. For a stability-indicating method that will run over weeks and months, source contamination isn’t a cosmetic issue. It’s a data integrity problem.
For the MRM transitions: the peptide carries two arginine residues with high proton affinity. In positive ESI mode, it readily forms a doubly charged ion [M+2H]²⁺ at m/z 445.5. That’s the precursor. The primary product ion is m/z 174.1, the characteristic immonium ion from the arginine side chain. High intensity, high selectivity.
| Parameter | Value | Rationale |
|---|---|---|
| Ionization mode | ESI positive | Arginine side chains have strong proton affinity |
| Precursor (Q1) | m/z 445.5 | [M+2H]²⁺, most stable and intense charge state |
| Product ion (Q3) | m/z 174.1 | Arginine immonium ion, characteristic and selective |
| Capillary voltage | 3.5 kV | Optimal droplet formation in high-organic HILIC solvent |
| Desolvation temp | 600°C | Necessary to flash-evaporate cosmetic polymer residues |
I didn’t get these from auto-tune alone. The precursor came from the molecular weight and charge state logic. The product ion came from fragmentation pathway analysis of the sequence. Auto-tune confirmed and refined. It didn’t discover.
P.S. Those table values were my starting parameters. They worked, but I saw slight tailing. I’ll write up the final method in a future piece.
On the CoA shortcut
A shortcut you’ll see proposed is weight balance: substantiate the peptide concentration from the raw material vendor’s CoA and skip testing the finished product. Just do the math on what went in.
It isn’t analytically defensible. The CoA tells you what the supplier claims went into the drum. It says nothing about what survived formulation, what degraded during processing, or what the finished product actually contains.
Regulators are closing that door anyway. In 2026 China’s NMPA released new cosmetic standards that specify direct testing requirements for actives like Acetyl Hexapeptide-8. Brands that filed on paper compliance are now looking at rejections and delays. The shortcut only looks cheaper until the file comes back.
What this approach actually delivers
The efficiency gains are real because every experiment was designed from chemical logic rather than guesswork.
I didn’t run 30 column-pH-gradient combinations and pick the best one. I predicted which pH would shift the glutamic acid ionization state, worked out which column type would separate the peptide from the matrix, and determined which mobile phase composition would maximize ESI sensitivity. Then I ran experiments to confirm and refine. The instrument time I used was the minimum needed to validate what I already expected to be true.
The whole chain, settled on paper before the first injection:
flowchart TD
A[Start with the structure] --> B[Map logP, pKa,<br/>ionizable groups]
B --> C{How polar?}
C -->|"Very polar (logP −6.3)"| D[C18 fails: elutes at the<br/>front, in the matrix noise]
D --> E[HILIC: polar retains late,<br/>in a clean window]
E --> F[Mobile-phase pH ~5:<br/>deprotonate the Glu, shift retention]
F --> G[High-organic mobile phase:<br/>stronger ESI]
G --> H[MRM 445.5 → 174.1<br/>from MW + charge logic]
H --> I[Bench time only to<br/>confirm and refine]The speed comes from deciding more at the desk and less at the bench.
A stability-indicating method like this runs for the product’s full shelf-life testing: accelerated stability at 40°C/75% RH, real-time at warehouse conditions, and in-use simulation. The in-use protocol (open the container, add a finger, close it, run it daily for the shelf life) caught variability the accelerated conditions didn’t. That’s a separate design decision and worth its own piece.
Why this matters beyond one molecule
Most SME founders and indie brand operators I talk to are at the mercy of whatever their lab runs by default.
Labs run what you order. They don’t tell you whether the test is right for your matrix. They don’t flag when the standard protocol is going to miss the compound you’re actually trying to detect. They send you a report and you accept it because you don’t have 14 years of LC-MS experience to interrogate it.
The method logic, the instrument choice, the sample prep, the interpretation: that’s the part that isn’t in the report. That’s the part you’re paying for when you hire an analytical scientist instead of just a lab.
If you’re developing a cosmetic, supplement, or functional food with an active ingredient that needs analytical verification, the question to ask isn’t “which lab.” It’s “what should the method actually be doing, and does the person I’m talking to know the answer before they start running samples.”
Key References
- Safety Assessment of Acetyl Hexapeptide-8 Amide as Used in Cosmetics, Link
- Five Cosmetic Industry Standards Released, Covering Technical Requirements for Biotechnological and Plant-Based Ingredients | News | GlobalCosing, Link
- Acetyl Hexapeptide-8 in Cosmeceuticals—A Review of Skin Permeability and Efficacy, Link
- Quantitation of Acetyl Hexapeptide-8 in Cosmetics by Hydrophilic Interaction Liquid Chromatography Coupled to Photo Diode Array Detection - MDPI, Link
- What are Matrix Effect in Liquid Chromatography Mass Spectrometry? | NorthEast BioLab, Link
- China: NMPA Updates Cosmetic Safety Technical Standards (STSC 2015) - Complife Group, Link
- Comparison of Multi-Mode Scherzo SS-C18 and HILIC Mode Column in the Retention, Link
- Applications of Hydrophilic Interaction Chromatography in Pharmaceutical Impurity Profiling: A Comprehensive Review of Two Decades - MDPI, Link
- Stability Programs: A Guide to Design, Data & Shelf Life | IntuitionLabs, Link