A fronting peak cost me 2 days once.
It was a UPLC method, bioequivalence work. The front edge of every peak leaned forward and would not sharpen. So I did what most people do. I blamed the column and changed it. Same lean. I re-prepped every sample. Same lean. Then I started to suspect the instrument.
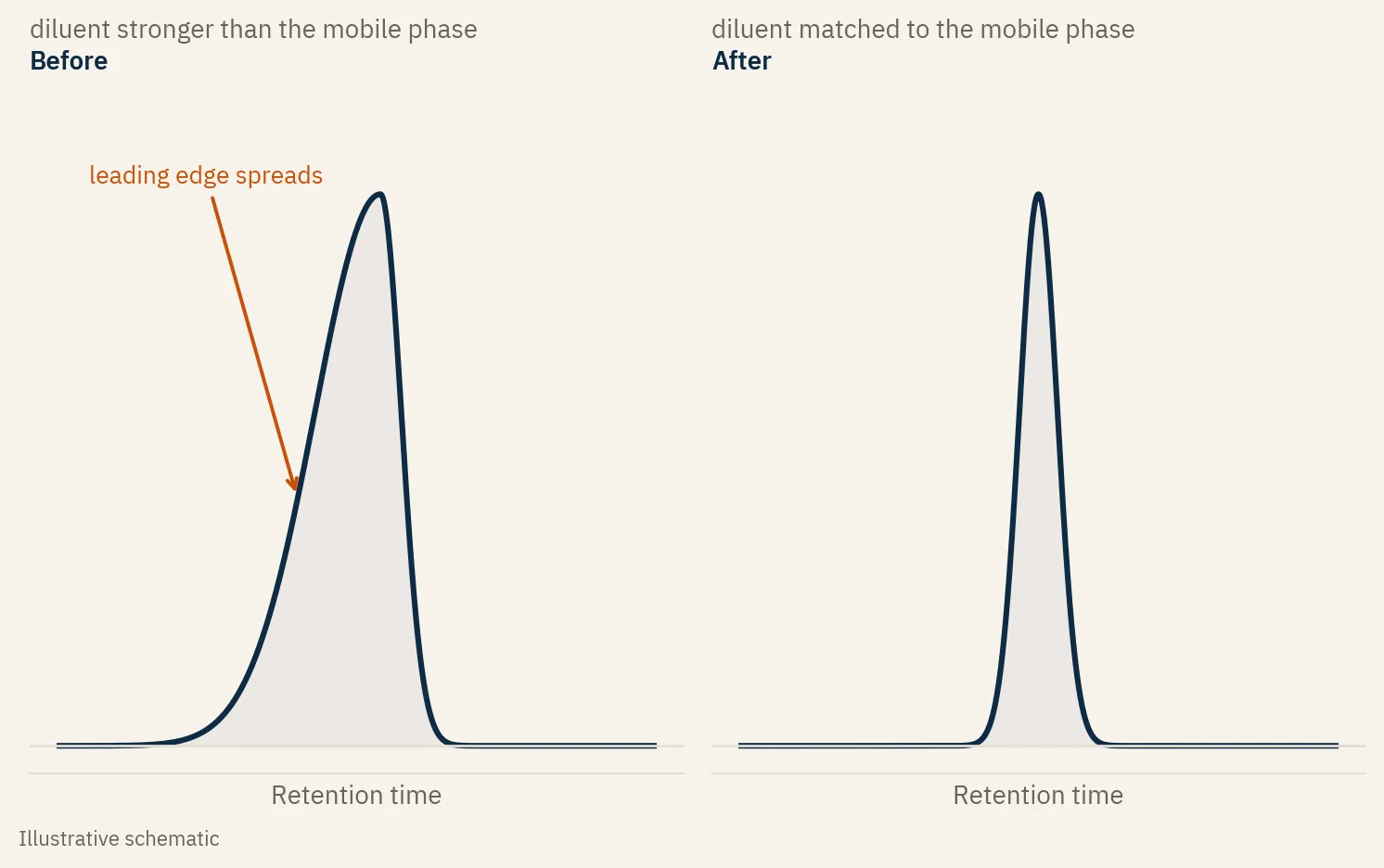
The column was fine. The instrument was fine. My diluent was stronger than my mobile phase.
The sample sat in a solvent stronger than the mobile phase it entered. At the head of the column the band could not focus, so the front of the peak spread before the column could pull it back. I weakened the diluent, closer to the starting mobile phase, and the next injection was clean.
2 days of work, for something a 30-second check would have caught.
Peak shape is a sample problem first
Peak shape gets blamed on the column or the instrument all the time. Those are the expensive things in the room, so that is where the eye goes. But the cause is usually upstream, in the sample: the diluent, the injection solvent, how much you put on the column.
That is not a comfortable place to look. It feels too simple to be the answer. So we skip it, swap the column, and lose two days proving the column was never the problem.
What fronting actually is
A symmetric peak rises and falls at the same rate. A fronting peak has a long, shallow leading edge and a sharper back. The front leans out toward the start of the run.
Splitting is the same problem one step worse. When the mismatch is bad enough, the band breaks into two and you get a shoulder or a double peak. Same cause, louder symptom.
The strong solvent effect
Here is the mechanism in one line. If your sample is dissolved in a solvent stronger than the starting mobile phase, the band does not focus at the head of the column.
In reversed phase, stronger means more organic. The analyte wants to move with that strong plug of solvent instead of sticking to the column and waiting for the gradient. Part of the band runs ahead. That is the leading edge you see. Weaken the diluent and the band focuses tight before it travels, and the peak comes back.
Where to look, cheapest first
Work the list in order. The first one is the most common and the cheapest to fix. The last one is the only case where the column is actually at fault, which is why it is last.
| Cause | The tell | Confirm it | Fix |
|---|---|---|---|
| Diluent stronger than the mobile phase | Fronting on most peaks, worse for early eluters | Dissolve the sample in mobile phase and re-inject | Weaken the diluent to match or sit below the starting mobile phase |
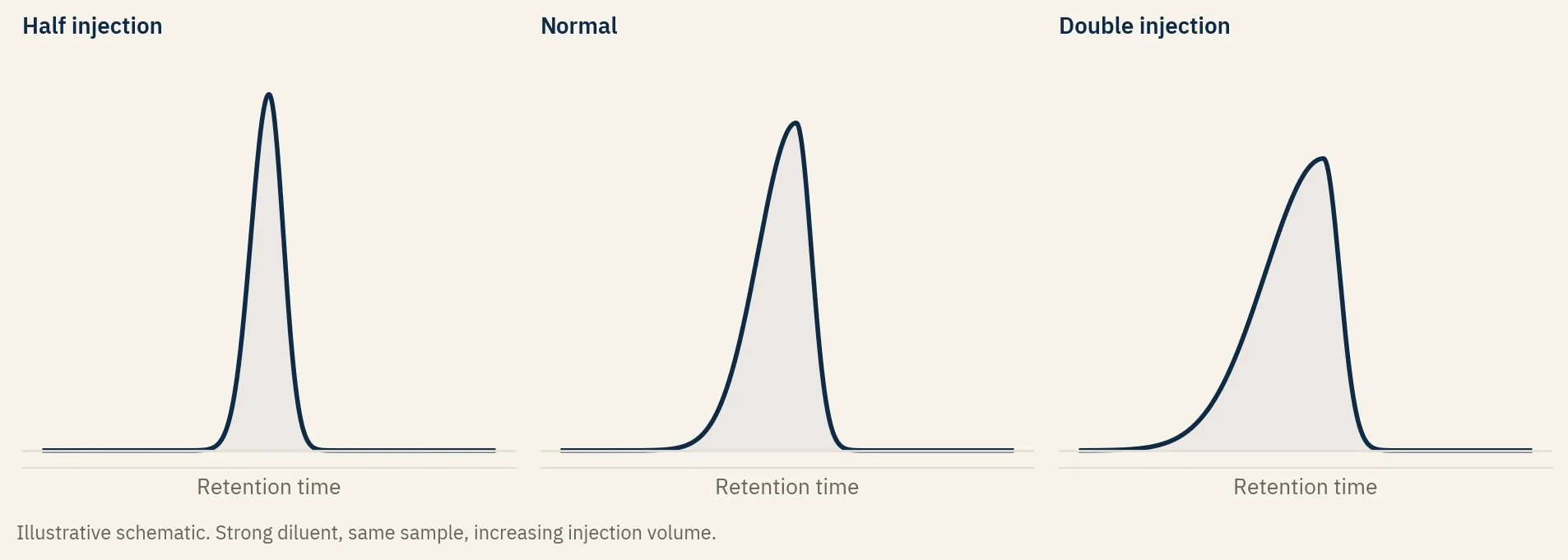
| Injection volume or concentration too high | Fronting that grows with how much you inject | Halve the injection volume | Inject less, or dilute the sample |
| Sample overload, too much mass on the column | Fronting on the major component, retention drifts with load | Inject a more dilute standard | Reduce the on-column mass |
| Column void or bed collapse | Splitting or fronting that does not move with diluent or volume, and gets worse as the column ages | Run the tests above and watch the shape stay put | Replace the column. This is the real-column case |
The confirm test
You do not have to guess which one it is. Change the injection volume and watch.
Halve it, then double it. If the peak sharpens when you inject less and worsens when you inject more, you are looking at a solvent or load effect, not the column. The distortion scales with how much strong solvent you put on the head of the column.
If the shape does not move with volume at all, the cause is somewhere else, and now the column is worth a serious look.
The diluent rule
The fix for the most common case is simple. Match your diluent to the starting mobile phase, or make it weaker.
A rule of thumb that has saved me a lot of trouble: in reversed phase, make the injection solvent about 10 percent more aqueous than the starting mobile phase composition. Weaker than the start gives you margin, and the band focuses instead of spreading.
The one thing that stops you is solubility. If the analyte will not stay in solution at that aqueous fraction, you cannot just weaken the diluent. Then you reduce the injection volume or the concentration instead, which gets you a clean peak when the chemistry will not let you change the solvent. One fixes the cause. The other manages around it.
One more, for ionizable and zwitterionic compounds. Keep the diluent and the mobile phase on the same side of the analyte’s pKa, or its isoelectric point if it is a zwitterion like a peptide or an amino acid. If they land on opposite sides, the molecule’s charge flips between the vial and the column, and the peak distorts for that reason alone. A strongly buffered mobile phase masks some of it. Designing the mismatch out is cleaner.
When it really is the column
Sometimes it is the column. A void at the head, or a collapsing bed, distorts peaks too, and it tends to throw splitting as readily as fronting.
You tell it apart by what does not change. If you weaken the diluent and cut the injection volume and the shape stays bad, the sample side is not the problem. A column that has been drifting worse over weeks, with high backpressure or a known injection count, points the same way. Reverse-flush it or replace it.
The point is the order. The column is the last thing you check, not the first, because it is the most expensive to swap and the least often guilty.
A method that only behaves on one system
There is a version of this that bites later. You tune and tune until the peak looks perfect, then the method moves to another instrument and the fronting comes back.
If a method only behaves after instrument-specific tuning, it is not robust. It is locally optimized, and the tuning hid the cause instead of fixing it. For gradient methods the usual culprit on transfer is dwell volume: a different system delivers the gradient to the column at a different time, selectivity shifts, and a method tuned to one dwell volume will not hold on another. Fix the actual cause during development, account for dwell volume, and the method travels. I wrote more about building this in early in method development as risk mitigation.
Before you blame the hardware
The next time a peak fronts and the obvious fixes do not work, stop before you swap the column. Compare your diluent to your mobile phase. Halve the injection volume and look. The cause has been simple and documented for decades. We just keep rediscovering it.
Questions about any of this? You can reach me at hello@nalam.ca or on LinkedIn.
Further reading
- Dwight Stoll and John Dolan, the “LC Troubleshooting” column in LCGC, archived at the LC Troubleshooting Bible. Dolan’s columns on injection-solvent effects are the canonical reference for this problem.
- L.R. Snyder, J.J. Kirkland, J.W. Dolan, Introduction to Modern Liquid Chromatography (Wiley). The standard text for method development.